Materials/chemicals have broad and deep impact on our lives. We are wearing hydrophobic fabrics that prevent us from wetting in rain. The current information/computer industries rely on the silicon-based semiconducting materials. Extensive efforts are made to make energetic materials which can replace petroleum and solar cells with low efficiencies. Pharmaceutical companies strive to discover drugs that can cure diseases.
Discovery of the materials/chemicals best suited for solving a given problem is challenging. However, experimental screening of candidate materials/chemicals are often time-consuming and expensive. Fortunately, with the remarkably fast advances in computer hardware/sortware, we now can simulate the molecular details underlying various phenomena which are interesting to researchers in materials science and engineering, nanotechnology and engineering, chemistry, physics, and biology. Nobel Chemistry Prize in 2013 was awarded to chemists who developed molecular modeling software to simulate chemistry related problems.
In Computational Nanochemistry Lab (CNCL) at Pusan National University, we utilize computer simulation to design materials/chemicals for applications in nanotechnology, biomimetics, and energy. Specifically, we are interested in designing materials/chemicals related to superhydrophobic surfaces, water-resistant universal glue materials, and nanostructured thin organic films. A true innovation comes from understanding the fundamental science underlying the design of materials/chemicals. We therefore investigate the chemistry and physics of these applications as well.
We utilize various computer simulation methods including the first principles density functional theory (DFT) calculations, ab-initio molecular dynamics (AIMD) simulations, molecular dynamics (MD) and Monte Carlo (MC) simulations, Brownian dynamics (BD) simulations, and the lattice Boltzmann method (LBM). We also utilize the statistical mechanical/thermodynamic theories to tackle these problems. The current research interests of CNCL are following.
Interfacial Water Probed by Sum Frequency Generation Spectroscopy
Structurally ordered layers of water are universally formed on a solid surface in aqueous solution or under ambient conditions. Although such hydration layers are commonly probed via atomic force microscopy (AFM), the current understanding on how the hydration layers manifest themselves in an AFM experiment is far from complete. The sum frequency generation (SFG) spectroscopy can overcome the limits of probing hydration layer by AFM.
The high interface selectivity in the SFG spectrum arises from its centrosymmetric properties, which
are a consequence of computing the imaginary part for 2nd order susceptibility. This susceptibility is derived from the molecular polarizability tensor and dipole moment. As a result, the orientation of water molecules, the chemical bonding, especially hydrogen bonding, and the stretching of OH bond of water molecules can be probed by SFG spectrum. However, still the expensive computation cost is remaining as challenging problem to obtain SFG spectrum.
In our group, we study the behavior of water molecules in hydration layer on various solid surfaces by applying the neural network potentials (NNP) based on the results of ab initio MD simulation to overcome the limitations of computation problem.
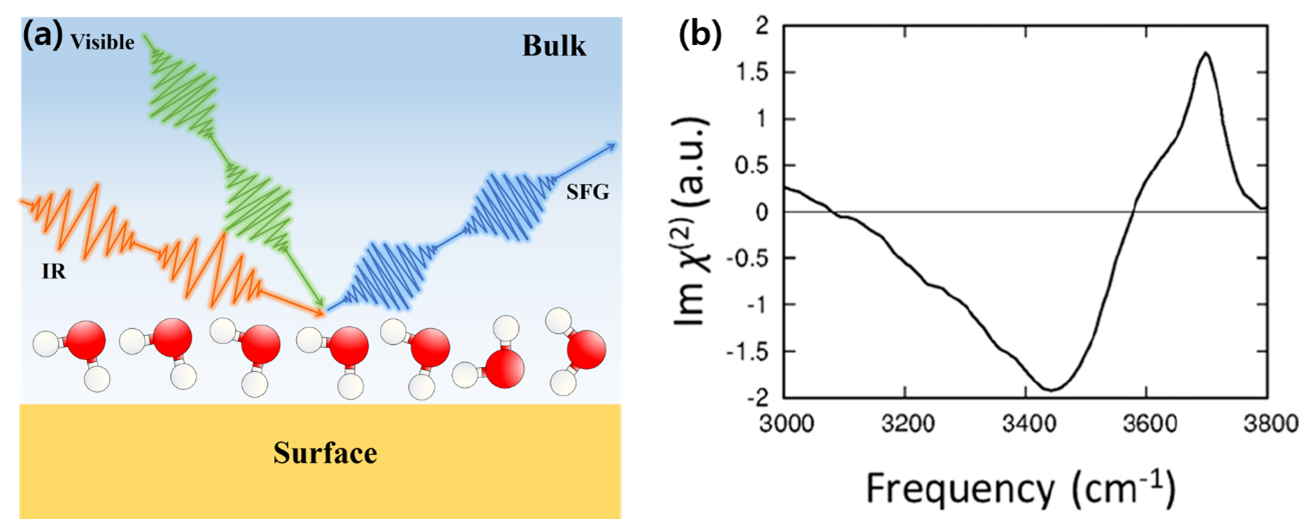
Figure
(a) Schematic of SFG spectrum analysis and (b) the example of the SFG spectrum of the interfacial water.
Computational Design and Development of New Energy Materials
Lithium-ion batteries (LIBs) are the preferred technology for electric vehicles, power tools, and portable electronics due to their high energy and power density. However, due to the scarcity of lithium and its high cost, the current LIB technology is unable to meet the large-scale global demand. Consequently, increasing their affordability and effectiveness can significantly broaden their applications and make new technologies possible that are not possible with conventional LIBs. Similar battery technologies, like potassium- or sodium-ion batteries, can be affordable substitutes for Li-based rechargeable batteries because of their low cost and widespread availability.
The main obstacle is the development of new electrode materials with high energy and power densities, extended cycle lives, and affordable prices. Since the properties of a material greatly influence its performance, electronic structure calculations can be used to design new energy materials.
Using a variety of computational tools, our group investigates the fundamental physics guiding the electrochemical behavior and performances of the new electrode materials for Li/Na/K ion batteries.
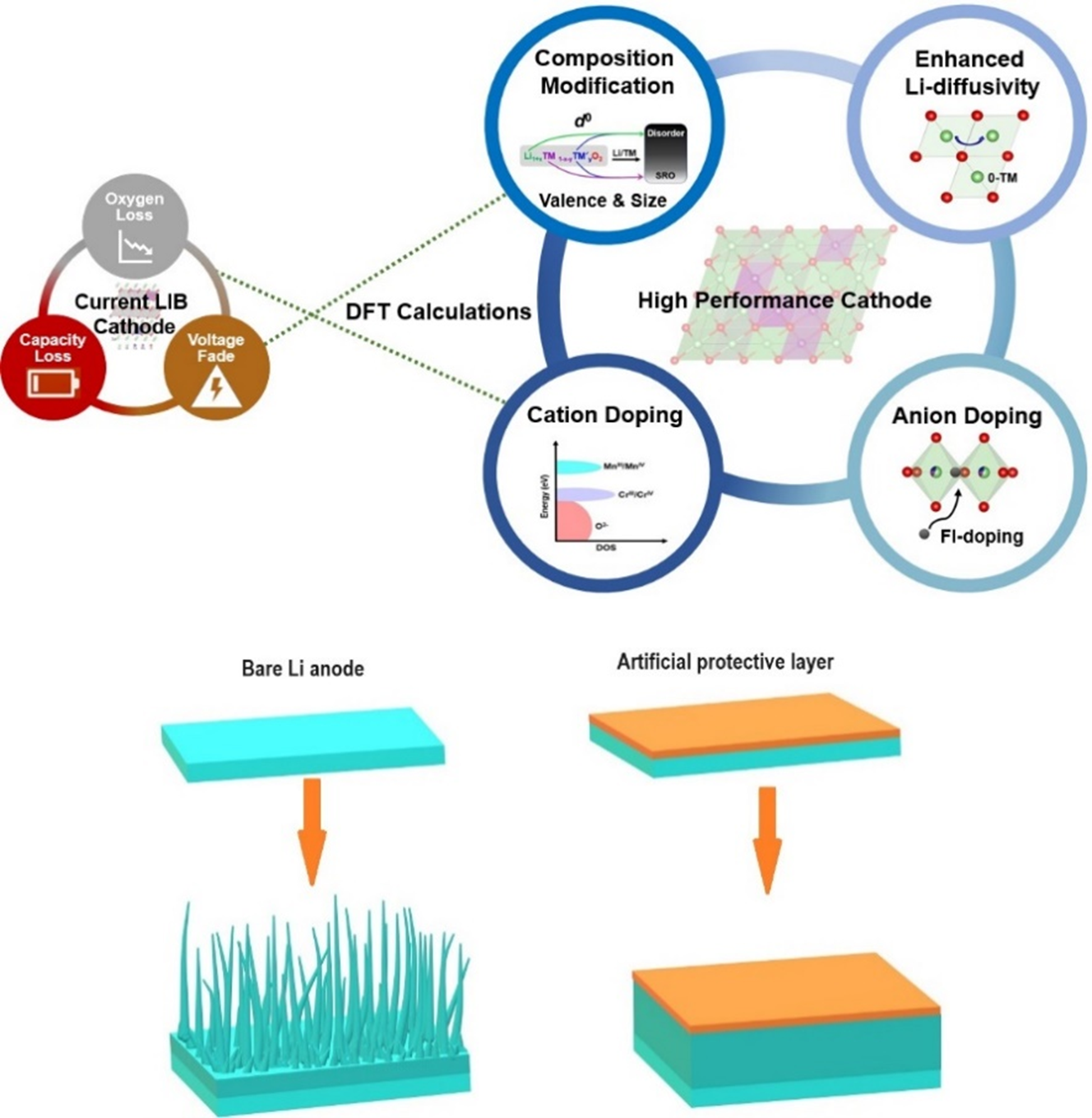
Figure
Schematic illustration of issues with current LIB cathodes and development of promising cathode materials for LIBs from theoretical screening and rational design (top) and suppression of Li-dendrite growth by artificial solid electrolyte interface (bottom).
Related articles
"Stabilizing a Nickel-Rich (LiNi0.89Co0.055Mn0.055O2) Cathode Material by Doping Zirconium or Molybdenum: A First-Principles Study" J. Phys. Chem. C v. 125(50), 27543-27555 (2021);
"Role of a Solid-Electrolyte Interphase in the Dendritic Electrodeposition of Lithium: A Brownian Dynamics Simulation Study" J. Phys. Chem. C v. 124(17), 9134-9141 (2020);
“First-principles study on the two-dimensional siligene (2D SiGe) as an anode material of an alkali metal ion battery” Comput. Mater. Sci. v. 165, 121-128 (2019).
Computational Investigation of Dye Sensitized Solar Cells
In dye sensitized solar cells (DSSC), the sensitizer has an important role which absorbs the sunlight and transfers the electrons to the semiconductor from its excited state. Popular Ruthenium complexes such as N3 (red dye) and N749 (black dye) have achieved power conversion efficiencies over 11%. To improve the efficiency of DSSC, profound knowledge and a deeper understanding of the sensitizer is essential. Our research focuses on the design (molecular engineering) and characterization of organic and metal-organic sensitizers using the DFT and TDDFT quantum chemical simulations. The electronic structure, electrochemical, and optical properties of the sensitizers are systematically investigated computationally. We have theoretically investigated the effect of cyclometalation on the near-infrared absorption of a ruthenium sensitizer and addressed the importance of cyclometalation.
The simulation of dye adsorbed onto the semiconductor surface is one of the challenging tasks in understanding the mechanism of DSSC. Since the electronic coupling between excited state dye and semiconductor greatly depends on dye adsorption, we focused our research on the adsorption studies. The adsorption of the dye also can influence the conduction band of the semiconductor and thereby the open circuit voltage of DSSC. Using the first principles simulations we demonstrated the effect of acetonitrile solvent on the optical behavior of four cyclometalated ruthenium dyes and dye adsorbed TiO2 systems. All the dyes have shown more than two-fold hyperchromic shifts in the absorption upon solvation.
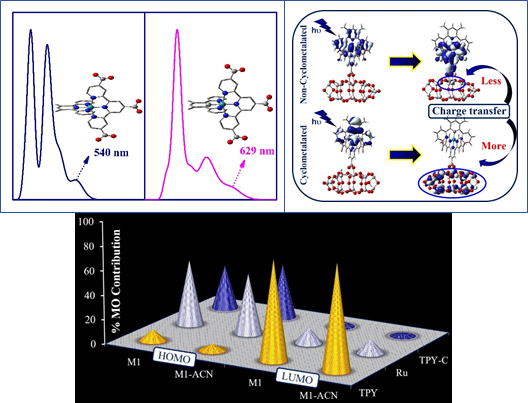
Figure
The effect of cyclometalation on the near-infrared absorption of a ruthenium sensitizer (top), and the partial molecular orbital contribution in frontier molecular orbitals of the ruthenium dye in vacuo and in acetonitrile (bottom).
Related articles
“Density functional theory study on ruthenium dyes and dye@TiO2 assemblies for dye sensitized solar cell applications” Sol. Energy v. 159, 283-290 (2018);
“Effects of Cyclometalation on the Panchromatic Ruthenium Sensitizer for DSSC Applications” Bull. Korean Chem. Soc. v. 38(10), 1209-1213 (2017).
Computer-Aided Drug Design
Computer-aided drug design (CADD) is a critical component of modern drug discovery and development. It is essential for making drug discovery more efficient, cost-effective, and targeted. CADD has made major contributions to help to bring compounds to clinical use. Indeed, several marketed drugs such as imatinib, zanamivir, nelfinavir, and other clinical candidates have been identified or optimized with the aid of molecular modeling techniques. Many computational methodologies are involved in the CADD technique, such as lead optimization, virtual screening, de novo design, and virtual library design. Especially in the pharmaceutical industries and academic organizations, virtual screening became a valuable method in which hit discovery cannot be usually conducted through expensive technologies. In this viewpoint, our research mainly focused on the CADD of novel organic molecules with improved biological properties like binding affinity and ADMET (absorption, distribution, metabolism, excretion, and toxicity) by applying various computational techniques. We also utilize a computer-aided approach to design peptides mimicking the angiogenic activity of Visfatin, showcasing promising results in neovascularization. As we continue to explore their potential and identify suitable target receptors, the horizon of angiogenesis research beckons with transformative possibilities. These studies highlight the multidisciplinary nature of research in drug discovery, offering insights into potential therapeutic avenues and advancing our understanding of molecular interactions.
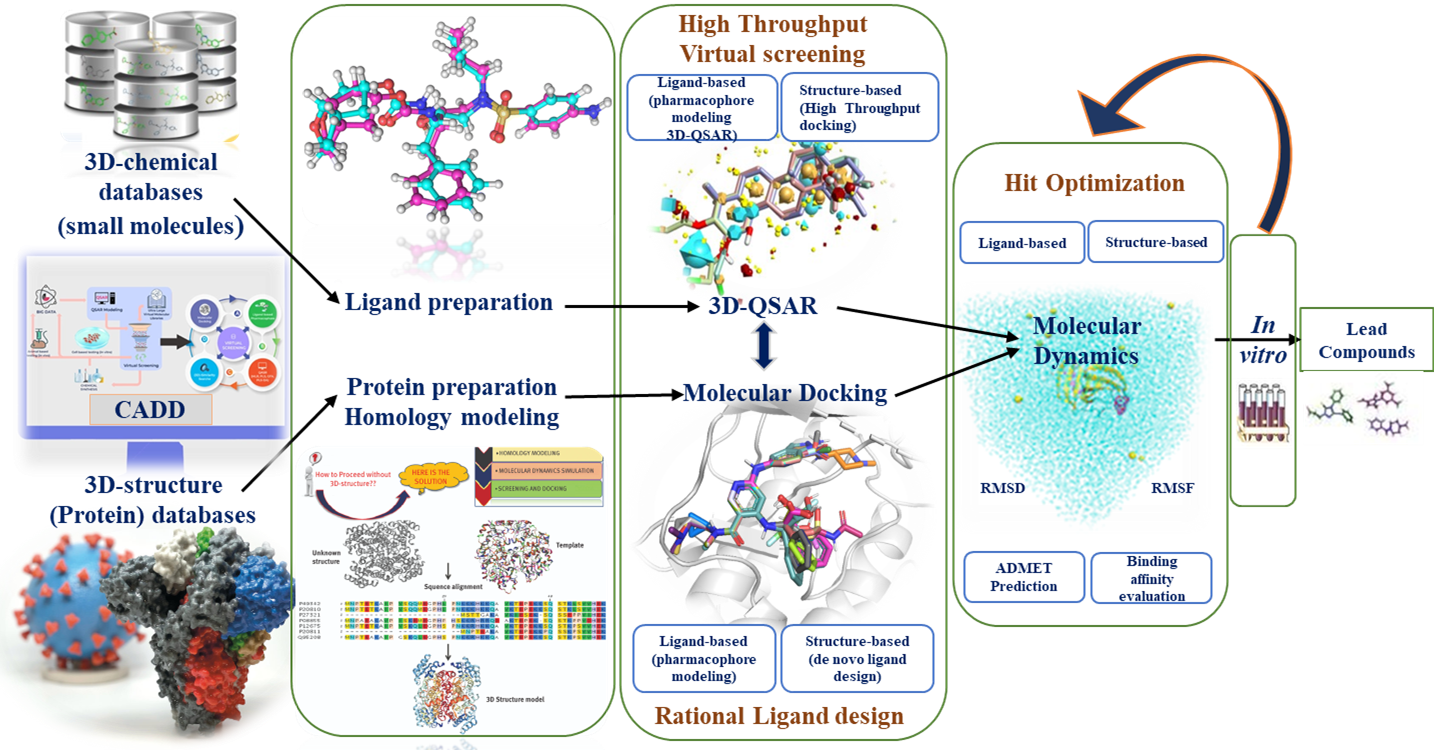
Figure
Schematic representation of the main steps involved in the computer-aided drug design and the applications of computational methods at different stages.
Related articles
“Computer simulation approach to the identification of visfatin-derived angiogenic peptides” PLOS One. v. 18 pp0287577 (2023).
“Development of Novel Chromones as Antioxidant COX2 Inhibitors: In vitro, QSAR, DFT, Molecular Docking, and Molecular Dynamics Studies” J. Bio Mol. Str. Dyn. (2023).
“A Combination of Pharmacophore-Based Virtual Screening, Structure-Based Lead Optimization, and DFT Study for the Identification of S. epidermidis TcaR Inhibitors" Pharmaceuticals. v.15, 635 (2022).
“Structure-based lead optimization to improve the antifungal potency of the tetrahydroimidazo pyridine inhibitors targeted to Candida albicans dihydrofolate reductase and lanosterol 14-alpha-demethylase” Med. Chem. Res. v. 28, 1674-1682 (2019).
Modeling and Application of Artificial Neural Network
An Artificial Neural Network (ANN) is a machine learning algorithm inspired by the biological neural network, which involves the synapses of neurons. It is used to estimate or approximate functions by translating a large number of inputs into a target output. This information processing technique operates in a manner similar to the human brain's processing of information.
ANNs are constructed from a series of layers, and each layer comprises many "neurons." Each neuron accepts an input value from the previous layer and maps it into a non-linear function. The output of this function is then used as the input for the next layer in the ANN. This process continues until it reaches the last layer, where the output corresponds to the objective to be predicted.
In computational chemistry, various ANN algorithms are utilized for Quantitative Structure-Property Relationship (QSPR), a method that seeks to establish models relating the chemical structural features of descriptors to their specific physical/chemical properties.
In our group, we get the relationship between surface structure and super-hydrophobicity through ANN from the simulated results using MC simulation and predict the contact angle for superhydrophobic surface for different types of surface geometry by using the convolutional neural network (CNN), an algorithm of ANN.
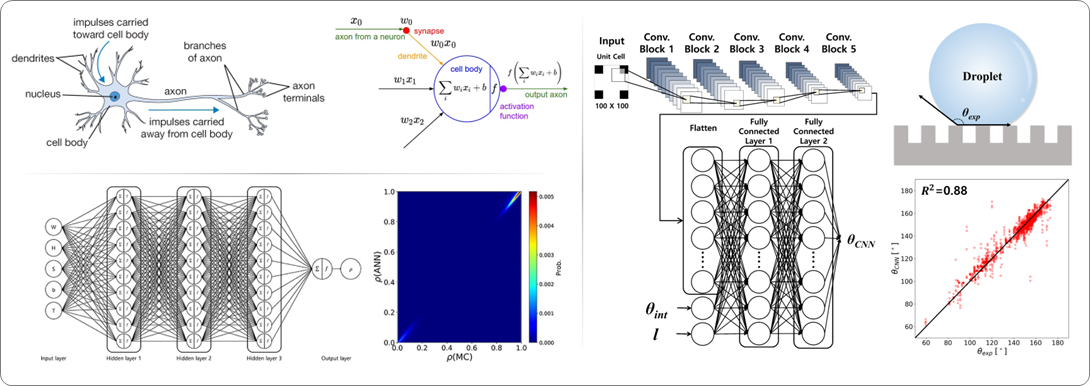
Figure
Structure of a typical biological neuron and artificial neuron (top left). The architecture of ANN (bottom left) and CNN (right) models for prediction super-hydrophobicity and contact angle, respectively.
Related articles
“Machine-Learning Approach in Prediction of the Wettability of a Surface Textured with Microscale Pillars” Langmuir, v.39, 17471-17479 (2023)
“Neural network modelling of the wettability of a surface grooved with the nanoscale pillar” Chem. Phys. Lett. v.768, 138360 (2021)
Biomimetic Water-resistant Glue Materials
A glue sticking irreversibly to wet surface has broad applications in areas, such as tissue adhesives and dental cements. Synthesizing a water-resistant glue is elusive however. Remarkably, marine mussels can adhere to virtually any surface under wet conditions, even under saline and tidal conditions ( see the figure below). Mussels contain an unusally high content of 3,4-dihydroxy-L-phenylalanine(DOPA).
Our lab works on computational design of glue materials that mimic the mussel adhesive proteins(MAPs). To do so, we study two aspects of the adhesion of mussel. First, the initial anchoring of MAP on a surface via DOPA functionalities. Second, a Fe(lll)-mediated curing process in which extensive cross-linking of these anchored MAPs occurs. The anchoring was studied by using DFT calculation for the adsorption of MAP on a silica surface (figure below). The cross-linking was studied by DFT calculation on the Fe(lll) coordination complex with DOPA ligands(figure below).
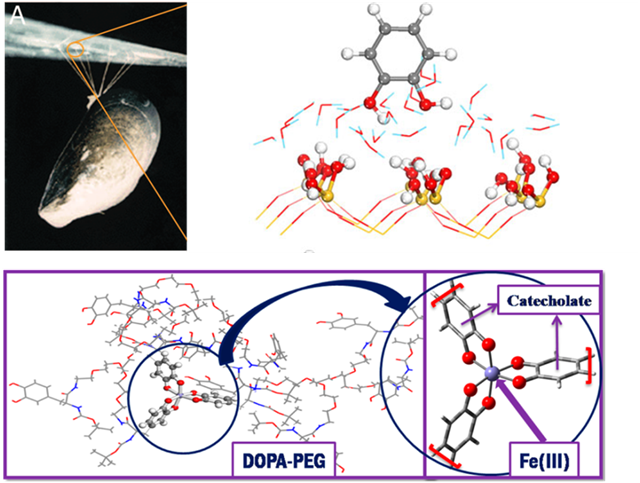
Figure
Mussel adhering to a surface through threads (top left). DFT simulation on the anchoring of DOPA on a silica surface covered with water (top right). DFT simulation on the crosslinking of DOPA modified polymer by forming the coordination complex with Fe(lll).
Related articles
"A density functional theory study on the underwater adhesion of catechol onto a graphite surface" Phys. Chem. Chem. Phys. v. 23(2), 1031-1037 (2021);
“Density Functional Theory Study on the Cross-Linking of Mussel Adhesive Proteins” J. Phys. Chem. B v. 119(17), 5496-5505 (2015);
“Adsorption of catechol on a wet silica surface: density functional theory study” Theo. Chem. Acc. v. 130(2-3) 333-339 (2011);
“Density Functional Theory Study of Catechol Adhesion on Silica Surfaces” J. Phys. Chem. C v. 114(48), 20793-20800 (2010).
Wettabilities of Surfaces
Superhydrophobic surfaces have wide applications, such as water harvesting, impermeable textiles, antifogging, and self-cleaning materials(e.g., paint and windows). The most venerable superhydrophobic surface is a lotus leaf which is texturized with micropillars (see figure below). A water droplet on a pillared surface exists in either Wenzel or Cassie state, depending on whether it is impaled by the pillars or not. A pillared surface that preferentially induces a Cassie state is desirable for an enhanced hydrophobicity.
Precise control of the size, shape, and pitch of the nano- or micro- pillars is possible with the assistance of micro- and nano-electromechanical(MEMS and NEMS) technologies. Nevertheless, experimental finding of an optimal pillared surface by varying the surface geometry can be both time-consuming and expensive. In this viewpoint, we use MD/MC simulation for screening the optimal surface geometries and thus providing guidelines for designing the optimal hydrophobic surfaces.

Figure
Water droplet on a lotus leaf (top left). Image of micro pillars on a lotus leaf (top right). Wenzel (bottom left) and Cassie (bottom right) state of a water droplet on a pillared surface.
Related articles
“Wetting Transition of a Cylindrical Cavity Engraved on a Hydrophobic Surface” J. Phys. Chem. C v. 122(4), 2122-2126 (2018);
“Contrasting water adhesion strengths of hydrophobic surfaces engraved with hierarchical grooves: lotus leaf and rose petal effects” Nanoscale v. 9(42), 16200-16204 (2017);
“Molecular Dynamics Study of the Hydrophilic-to-Hydrophobic Switching in the Wettability of a Gold Surface Corrugated with Spherical Cavities” Langmuir v. 32(37), 9658-9663 (2016);
“Monte Carlo Study on the Wetting Behavior of a Surface Texturized with Domed Pillars” J. Phys. Chem. C v. 118(45), 26070–26079 (2014);
“Molecular dynamics study on the wettability of a hydrophobic surface textured with nanoscale pillars” Phys. Chem. Chem. Phys. v. 16(12), 5613–5621 (2014);
“Drying Transition of Water Confined between Hydrophobic Pillars” J. Phys. Chem. C, v. 116(36), 19233–19239 (2012).
Phase Transitions of Confined Water
When water vapors are confined between solid walls in pores, their properties are significantly different from those of the bulk phase. A liquid water condenses at a pressure significantly lower than the bulk saturation value when it is confined between hydrophilic walls. This is called capillary condensation. In the case of hydrophobic walls, a drying transition occurs where liquid water evaporates at a pressure above the bulk liquid-vapor transition pressure.
The phase behavior of confined water is very important in many applications. Particularly, we are interested in the capillary condensation/drying in AFM experiments. An AFM tip with an apex on the nanometer scale is used widely for imaging and nanocontact printing. In these processes, the ambient water vapor condenses spontaneously into a liquid meniscus, which bridges the tip and surface. The nascent meniscus exerts an adhesion force (typically, several nN in magnitude), and this capillary force dominates over the van der Waals and electrostatic forces between the tip and surface. Therefore, it is important to understand the capillary force to interpret AFM images. The drying transition of water confined between hydrophobic objects was ascribed to the driving force in the self assemblies of proteins and carbon nanotubes.
Our group utilizes MD/MC simulation to study the phase behavior of water confined in nano- or micro- pores. We investigate how the phase depends on relative humidity, the geometry of pore, and surface energy of solid.
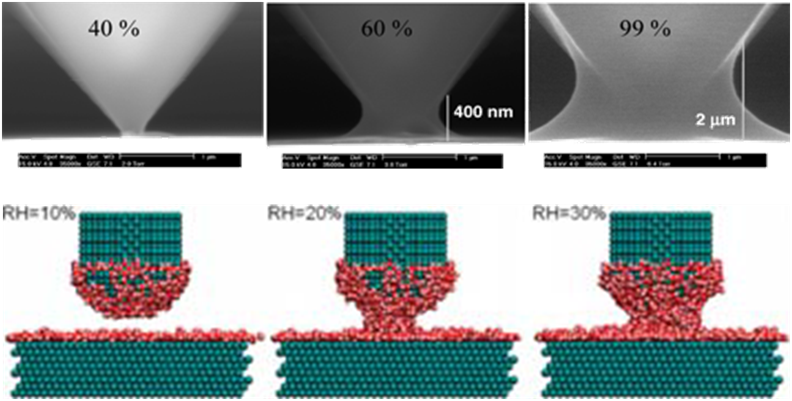
Figure
Experimental (top) and MD simulation (bottom) images of water condensed between an AFM tip and a surface at various relative humidities.
Related articles
“Monte Carlo Study on the Water Meniscus Condensation and Capillary Force in Atomic Force Microscopy” J. Phys. Chem. C v. 116(41), 21923–21931 (2012);
“Lattice Gas Monte Carlo Simulation of Capillary Forces in Atomic Force Microscopy” J. Adhes. Sci. Tech. v. 24(15-16), 2429–2451(2010).;
“Molecular simulation of the water meniscus in dip-pen nanolithography” Scanning v. 32(1), 2–8 (2010);
“Molecular simulation of the nanoscale water confined between an atomic force microscope tip and a surface”, Mol. Sim. v. 35(6), 466–472 (2009).
Computational Study of Electron-Hole Recombination in Light-Emitting Diode(LED)
The next generation of display technology has been deemed possible by Micro-LEDs (µLEDs) because of their high brightness and ambient contrast ratio. Red, green, and blue LEDs are made possible by group III-nitride semiconductors, whose light emission spans the whole visible spectrum. For full-color displays, the µLED display technology needs individual µLED chips that are red, green, and blue (RGB). While efficient InGaN-based blue and green LEDs have been achieved, there are significant technological obstacles in the way of InGaN-based red LED implementation.
The high indium requirement of the InGaN-based red LED leads to a high defect density in the epi-layers because of the significant lattice mismatch between the InN and GaN. Furthermore, the high indium content produces a strong quantum-confined Stark effect (QCSE) and large piezoelectric fields. These phenomena reduce the spontaneous electron-hole recombination rate, which impairs device performance and internal quantum efficiency (IQE).
In our group, we use DFT simulation to investigate electron-hole recombination rates and thermal stability of InGaN-based LED materials.
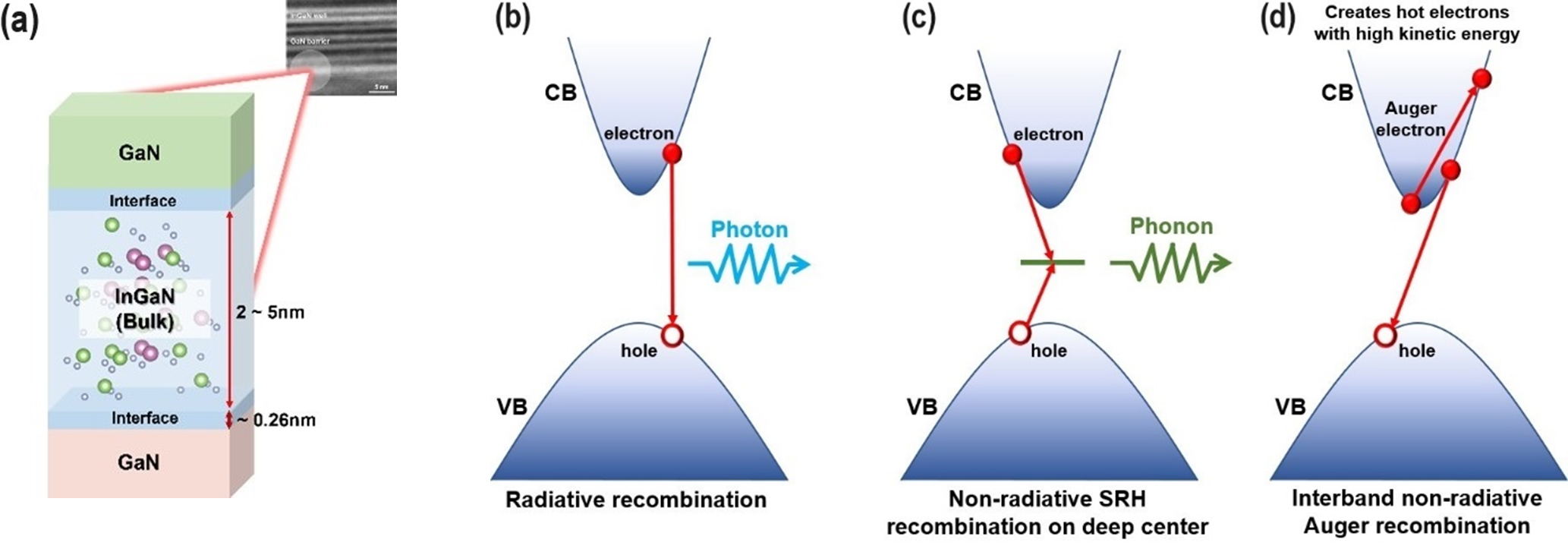
Figure
Schematic of InGaN quantum well on GaN substrate (a) and the main recombination mechanisms in the active quantum wells: radiative recombination by emitting photons (b); non-radiative Shockley-Read-Hall (SRH) recombination at deep centers (c); and direct inter-band Auger effect in semiconductors: an electron-hole pair recombines by exciting another electron to high kinetic energy (d).
Related Articles
“Recent progress of InGaN-based red light emitting diodes” Micro Nanostructures v. 183, 207669 (2023)
“InGaN-based red light-emitting diodes: from traditional to micro-LEDs” Jpn. J. Appl. Phys. v. 61, SA0809 (2022)
Computational Investigation of Perovskite Solar Cells
Perovskite solar cells (PSCs) have emerged as a promising avenue for the renewable energy landscape due to their potential for high efficiency, low cost, and tunability. However, several challenges remain regarding their stability, efficiency limitations, and toxicity concerns. This makes computational investigations crucial for gaining insights into the fundamental mechanisms and advancing technology. The presence of lead raises concerns about environmental toxicity and device stability. Crown ethers with specific cavity sizes exhibit a strong affinity for metal cations like Pb²⁺ through host-guest complexation. This makes them exciting candidates for capturing lead released from degrading perovskite layers.
Hole-transporting materials (HTMs) play a crucial role in PSCs by extracting and transporting holes generated in the perovskite layer to the positive electrode. Our computational studies are dedicated to developing efficient and stable HTMs. We focus our research on designing thermally and photochemically stable HTMs with high hole mobility. We leverage advanced computational tools to strategically tailor molecular structures, fine-tuning their energy levels for maximized performance and efficiency.
Light-induced degradation remains a significant challenge hindering the commercialization of PSCs. Understanding the underlying mechanisms at the atomic and electronic level is crucial for addressing this critical issue. Here, we focus our research on understanding degradation pathways by identifying initial defect formation, studying defect migration and interaction, and analyzing the role of dopants and interfaces. Moreover, employing theoretical investigation techniques, we analyze the Raman and surface-enhanced Raman spectra of both perovskite and modified perovskite. This elucidates crucial insights into the structure and dynamics of this prototypical material, particularly in the context of PSC applications.
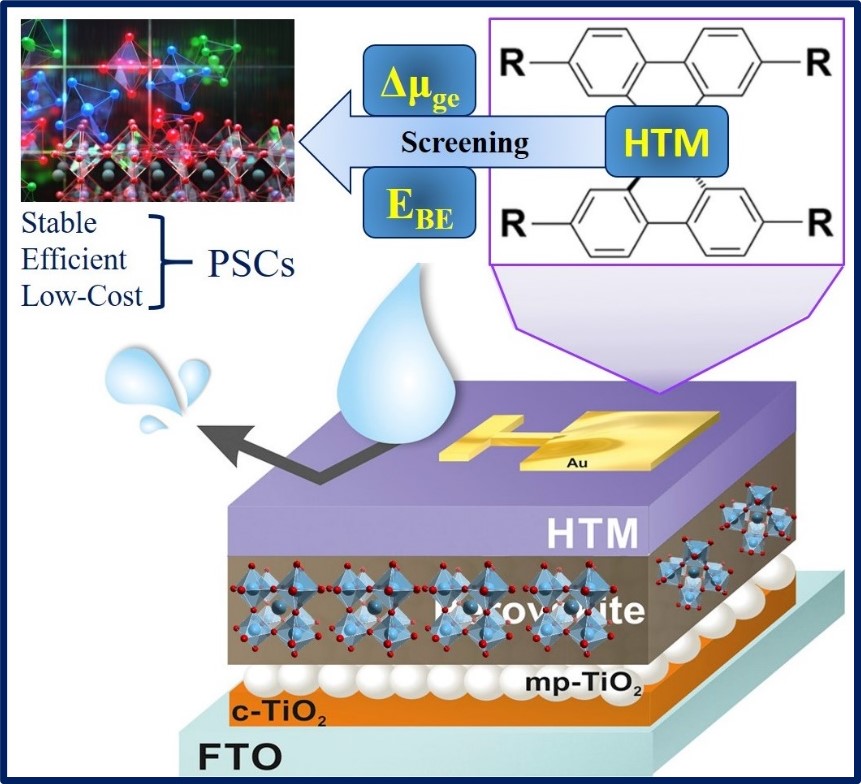
Figure
Screening and development of efficient and novel hole-transporting materials for perovskite solar cells.
Related Articles
“A Si-Substituted Spirobifluorene Hole-Transporting Material for Perovskite Solar Cells” ACS Energy Lett. v. 8, 5003-5011 (2023)
“Controlled Growth of Hybrid Halide Perovskites by Crown Ether Complexation for Perovskite Solar Cells” Helv. Chim. Acta. v. 106, e202200193 (2023)
Unraveling the Secrets of Hydrogen Evolution: A Journey with DFT Calculations
Hydrogen evolution offers immense potential as a clean and sustainable energy source. However, to fully unlock its potential, we need a deep understanding of the fundamental mechanisms at the atomic and electronic level. This is where computational study approaches powered by density functional theory (DFT) come into play.
Through precise DFT calculations, we can unravel the intricate relationship between intermediate adsorption energies, reaction pathways, and overall hydrogen evolution reaction (HER) performance in electrocatalysts. Investigating both the Tafel and Heyrovsky pathways is crucial for optimizing HER efficiency. We delve into these mechanisms at the atomic and electronic level by calculating the free energies of all relevant species (water, intermediates, hydrogen). This allows us to map the potential energy landscape of both pathways, revealing the transition states, potential barriers, and rate-determining steps for each.
By understanding these key factors, we focus on designing next-generation HER electrocatalysts with tailored intermediate adsorption energies, optimized reaction pathways, and enhanced catalytic activity. This journey with DFT calculations holds the key to unlocking the full potential of hydrogen evolution, paving the way for a cleaner and more sustainable future.
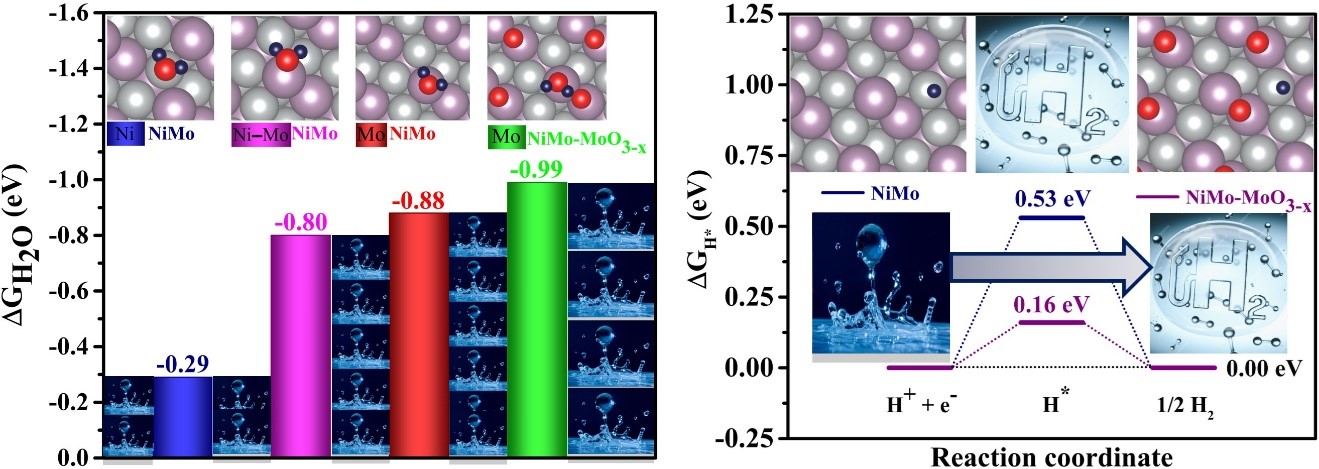
Figure
Adsorption free energies of water and hydrogen on electrocatalysts for hydrogen evolution reaction.
Related Articles
“Development of metal-organic framework-derived NiMo-MoO3-x porous nanorod for efficient electrocatalytic hydrogen evolution reactions” Appl. Catal. B: Environ. v. 328, 122421 (2023).